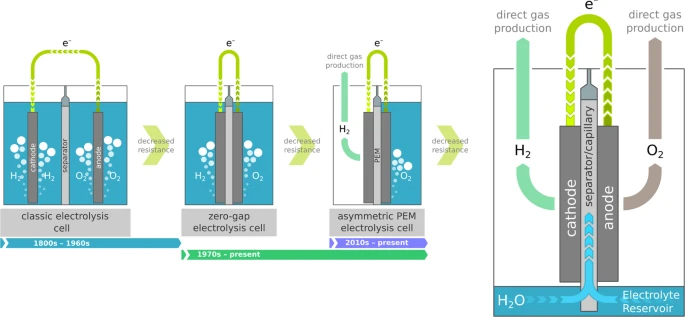
Renewable, or green, hydrogen will
play a critical role in the decarbonisation of hard-to-abate sectors
and will therefore be important in limiting global warming. However,
renewable hydrogen is not cost-competitive with fossil fuels, due to
the moderate energy efficiency and high capital costs of traditional
water electrolysers. Here a unique concept of water electrolysis is
introduced, wherein water is supplied to hydrogen- and oxygen-evolving
electrodes via capillary-induced transport along a porous
inter-electrode separator, leading to inherently bubble-free
operation at the electrodes. An alkaline capillary-fed electrolysis
cell of this type demonstrates water electrolysis performance
exceeding commercial electrolysis cells, with a cell voltage at
0.5 A cm−2 and 85 °C of only 1.51 V, equating to 98% energy
efficiency, with an energy consumption of 40.4 kWh/kg hydrogen (vs.
~47.5 kWh/kg in commercial electrolysis cells). High energy
efficiency, combined with the promise of a simplified
balance-of-plant, brings cost-competitive renewable hydrogen closer to
reality.
Introduction
Anthropogenic climate change, driven
largely by the burning of fossil fuels, poses a global existential
threat. This has motivated a growing number of nations and
corporations to aim for net-zero carbon emissions by 2050 to limit
global warming to 1.5 °C above pre-industrial levels1,2.
A critical element of the future net-zero
world will be renewable hydrogen, or green hydrogen, produced by water
electrolysis powered by renewable electricity, such as solar and wind.
The electrolysis of water requires the input of electrical energy and
heat energy, resulting in the evolution, from water, of hydrogen gas
at the cathode and oxygen gas at the anode according to:
2H2O(l)⇌2H2(g)+O2(g)E0=−1.229V
Green hydrogen will be essential
to the decarbonisation of hard-to-abate sectors such as steel
manufacture, long-haul transport, shipping and aviation1,2,3.
It may also be used for the seasonal storage of renewable electricity1,2,3,4,5,6,7
and as a chemical feedstock1,2,3,4,5.
However, the levelised cost of green hydrogen (LCOH) is presently not
competitive with fossil fuels. This is due to the high capital
expenditure (CAPEX) and high operational expenditure (OPEX) of
present-day water electrolysers. The OPEX is, by far, the larger
component of LCOH and it is dominated by the overall energy efficiency
of the water electrolyser and the cost of the input renewable
electricity to which it applies2.
At sub-MW scale, state-of-the-art commercial water electrolysers
typically require ~53 kWh of electricity to produce 1 kg of hydrogen,
which contains 39.4 kWh of energy, according to its higher heating
value (HHV)2.
Of that, the electrolysis cell, which is ~83% energy efficient (HHV)
at the operating current density, consumes ~47.5 kWh, with the
engineering system, known as the balance-of-plant, consuming the
remaining ~5.5 kWh2. The International Renewable Energy
Agency (IRENA) has set a 2050 target2
to decrease cell energy consumption to <42 kWh/kg. Any improvements in
net energy efficiency create a proportionally equivalent decrease in
the levelised cost of the produced hydrogen (Supplementary Fig. 1).
This work introduces a unique concept of
water electrolysis that promises notably reduced CAPEX and OPEX
compared to conventional water electrolysers, making renewable
hydrogen more cost-competitive with fossil fuels.
Inspired by the historic evolution of
water electrolysis cells, which recently culminated in asymmetric
polymer electrolyte membrane (PEM) cells that directly produce one of
the gases in a gas collection chamber rather than bubbling through the
liquid electrolyte8,
we have developed a capillary-fed electrolysis (CFE) cell concept in
which both gases are produced directly in gas collection chambers
(Fig. 1).
The aqueous electrolyte is constantly supplied to the electrodes by a
spontaneous capillary action in the porous, hydrophilic,
inter-electrode separator. The bottom end of the separator is dipped
in a reservoir, resulting in capillary-induced, upward, in-plane,
movement of electrolyte. Porous gas diffusion electrodes are held
against opposite sides of the separator, above the level of the
electrolyte. The electrodes draw in liquid laterally from the
separator and are covered with a thin layer of the electrolyte. The
application of sufficient voltage between the electrodes results in
the electrolysis of water, which is continuously replenished by water
moving up the separator from the reservoir. Because the generated
hydrogen and oxygen gases readily migrate through the thin layer of
liquid electrolyte covering their respective electrodes, the
capillary-fed cell concept provides for bubble-free electrolysis in
which water is converted directly to the bulk gases without forming
gas bubbles9,10,11,12,13.
Inspired by the historic evolution
of water electrolysis cell architectures culminating in the direct
production of one of the gases, the Capillary-Fed Electrolysis cell
directly produces both gases. Liquid electrolyte is continuously drawn
up the separator by a capillary effect, from a reservoir at the bottom
of the cell. The porous, hydrophilic separator sustains the flow rate
required for water electrolysis.
In so doing, the cell avoids bubbles
masking the electrodes and maintains access to the catalytic sites on
the electrodes. This includes access to the most active crevice, cleft
and defect sites that are the first to be blocked by bubble formation,
sometimes permanently so14.
It also ensures that water flow to an electrode does not counteract
gas flow away from the electrode, thereby avoiding the counter
multiphase flows inherent in conventional water electrolysers and
their associated mass transport limitations. In diminishing the energy
needed to overcome such inefficiencies, the capillary-fed cell
realises significantly improved energy efficiency.
It is also important to assess whether
new cell configurations increase or decrease the complexity (i.e. the
energy consumption and CAPEX) of the balance-of-plant. In the case of
CFE cells, notable simplifications in the balance-of-plant are
apparent. The absence of gas bubbles and associated gas-liquid froth
formation in the cell stack, removes the need for liquid circulation,
eliminating the gas-liquid separator tanks normally required and their
piping, pumps, and fittings (Supplementary Figs. 23).
The high energy efficiency, further, permits air-cooling, or radiative
self-cooling, eliminating need for water-cooled chillers
(Supplementary Tables 12).
The small volumes of liquid electrolyte in each cell reservoir
decrease the overall volume of water required (Supplementary Table 3).
Unwanted and wasteful shunt currents found in conventional alkaline
water electrolysers, may also be avoided. These simplifications in the
balance-of-plant lead to downward pressure on electrolyser CAPEX.
The capillary-induced flow of aqueous
27 wt% KOH electrolyte up a saturated, hydrophilic, porous, polyether
sulfone (PES) separator is initially measured and modelled,
demonstrating its ability to indefinitely support water electrolysis
at 1 A cm−2 and ≥80 °C for a height of up to 18 cm. This
height restriction, which is created by gravity, is taken into account
here, although it may in future be avoided by locating the reservoir
at the top of the separator.
In this work we show that a capillary-fed
cell, employing a known NiFeOOH oxygen evolution electrocatalyst on
the anode and Pt/C hydrogen evolution electrocatalyst on the cathode,
tested at 8085 °C with 27 wt% KOH electrolyte, yields water
electrolysis with performance that exceeds conventional, bubbled
control cells, and commercial alkaline and PEM cells. Faradaic
efficiencies approach 100%, with low gas crossover. Cell energy
efficiencies at 85 °C of 95% (HHV) at 0.8 A cm−2 and 100% (HHV)
at 0.3 A cm−2 (39.441.6 kWh kg−1 H2)
surpass the IRENA 2050 target and combine with the promise of a
simplified balance-of-plant to bring cost-competitive renewable
hydrogen closer to reality.
Results
Eq. (5)
permits a first-principles calculation of the capillary-induced rate
of in-plane transport of a liquid (e.g. an aqueous KOH electrolyte)
through a thin porous material using measurable or known quantities.
Capillary-induced, in-plane
transport of aqueous KOH electrolyte within porous polyether sulfone (PES)
filters
A search was undertaken to identify
potential inter-electrode separators that could draw up aqueous KOH
electrolyte by a capillary action. This led to a series of
commercially available porous, hydrophilic polyether sulfone
filtration membranes that were specified as having average pore
diameters of 0.45 μm, 1.2 μm, 5 μm, and 8 μm. Each was characterised
for its capacity to draw up a 27 wt% aqueous KOH solution from a
reservoir by capillary action. This electrolyte closely represents, at
2080 °C, the 6 M KOH that has historically been used in industrial
alkaline electrolysers. The capillary flow rates at different heights,
when the filter was full of liquid, were measured (as described in the
Method section and in Supplementary Fig. 4).
A linear flow regime, termed Darcy flow, is observed. A dry filter
filling itself for the first time exhibits non-linear Washburn flow
(Supplementary Fig. 4).
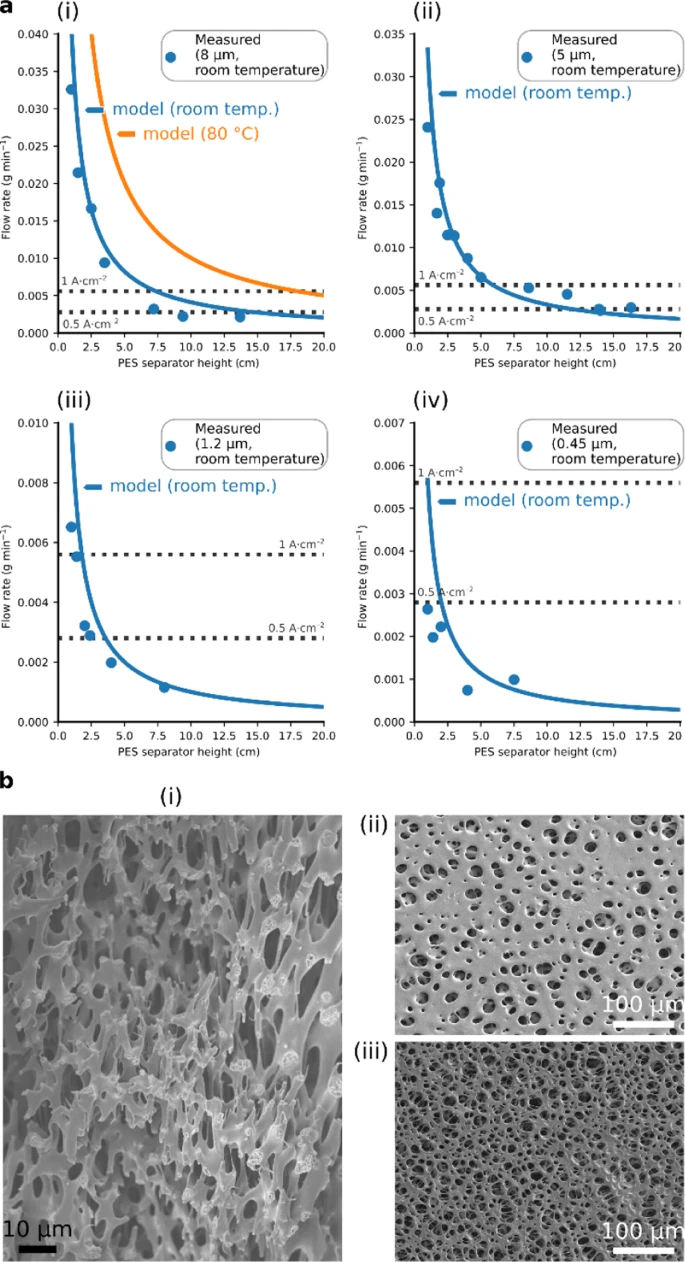
Figure 2a
shows the capillary-induced, in-plane, Darcy flow rates of the
polyether sulfone filters at room temperature. Modelled flow rates,
using Eq. (5),
showed reasonable agreement with the measured flow rates (Source
DataFigs. 24). The contact angle (70.3°) was measured with the
captive bubble technique; literature values provided surface tension
and viscosity16,17.
a Flow rates at different heights
inside a capillary-fed inter-electrode separator. Measured (data
points) and modelled (blue line) flow rates (Darcy flow), at room
temperature, of 27 wt% aqueous KOH within polyether sulfone (PES)
separators that were specified as having average pore diameters of: (i)
8 μm, (ii) 5 μm, (iii) 1.2 μm, and (iv) 0.45 μm. The measured data was
obtained as described in the Methods section and Supplementary Fig. 4.
The modelled data was obtained as described in the text. The orange
line in (i) depicts the modelled flow at 80 °C. The dotted lines show
the rate of water consumption by a 1 cm2 water electrolysis
cell operating at 0.5 A cm−2 (lower dotted line) and 1 A cm−2
(upper dotted line). The capillary-induced flow rate within the 8 μm
polyether sulfone separator is sufficient to supply water electrolysis
at 0.5 A cm−2 at a height of 15 cm at room temperature, and
at 1 A cm−2 at a height of 18 cm at 80 °C. b Pore
structure of the polyether sulfone separator. Scanning electron
micrographs of the polyether sulfone filter, showing: (i) its
structure in cross-section, and (ii) its gloss surface and (iii) its
matte surface.
The flow rates can be seen to decline
with increasing height and with smaller pores. The polyether sulfone
filter having 8 μm average pore diameter demonstrated the highest flow
rates (Fig. 2a(i))
and was selected for further use. If employed as an inter-electrode
separator, this filter has a modelled flow rate at room temperature
sufficient to indefinitely support capillary-fed water electrolysis at
0.5 A cm−2 up to a height of at least 15 cm (lower dotted
line in Fig. 2a(i)).
Modelling using Eq. (5),
indicated that at ≥80 °C, water electrolysis at 1 A cm−2
could be supported up to at least 18 cm in height (upper dotted line
in Fig. 2a(i)),
permitting the construction of water electrolysis cells with practical
height.
Pore structure, porosity, and the ionic
resistance of the polyether sulfone (PES) separator
Further investigations of the polyether
sulfone separator with 8 μm average pore diameter revealed it to have
a porous, open structure. Figure 2b
depicts SEM micrographs of its cross-section and two sides, one of
which had a gloss, and the other a matte appearance. It was found to
be 80% porous (see the Methods section).
The ionic resistance of the polyether
sulfone filter, when filled with 27 wt% KOH, was measured as described
in Supplementary Fig. 5.
Table 1
compares its ionic resistance at room temperature with those of
separators typically used in commercial alkaline (Zirfon PERL UTP 500)18
and PEM electrolysis cells (NafionTM 115 and NafionTM
117)19.
All of these ionic resistances include the resistance of electrolyte
incorporated within them. The polyether sulfone separator displayed
244 mΩ cm2 lower ionic resistance than Zirfon PERL UTP 500,
due to its lesser thickness and higher porosity. Its ionic resistance
was also 174 mΩ cm2 less than NafionTM 117 and
96 mΩ cm2 less than NafionTM 115. The low ionic
resistance of the PES separator compared to commercial separators
contributes to the high energy efficiency of the capillary-fed cell
described below.
Table 1 Ionic resistance of
separators at room temperature.
Capillary-fed electrolysis cell outperforms
conventional and commercial water electrolysis cells
To evaluate the CFE cell concept, a test
cell was fabricated using the polyether sulfone filter with 8 μm
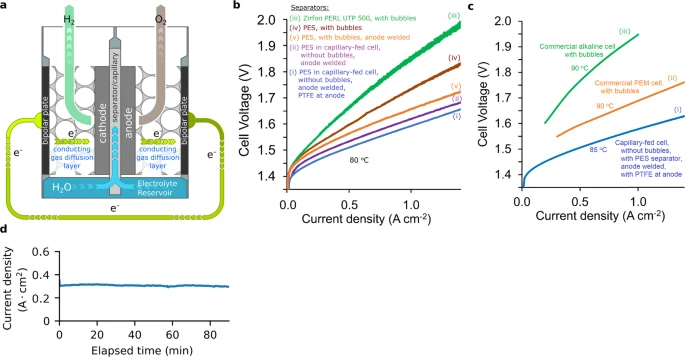
average pore diameter, as the electrode separator. Figure 3a
depicts a schematic of the cell. Polyether sulfone filters of much
smaller pore diameter have been previously tested as inter-electrode
separators in water electrolysis cells21,22.
a Schematic depiction showing how the
bipolar plate and conducting gas diffusion layer in the capillary-fed
cell were combined into a single bipolar plate structure that
comprised a sheet of Ni with many small holes to allow evolved gases
to exit the electrode. The anode electrode was welded to its bipolar
plate. The cathode was compressed against its bipolar plate and not
welded. Supplementary Fig. 6
provides a picture of the cell used. b Polarisation curves of
capillary-fed electrolysis cell and controls (80 °C). Plots of
(2-electrode) cell voltage vs. current density, from bipolar plate to
bipolar plate across the cell, excluding cathodic oxygen
depolarisation, of cells with identical NiFeOOH anodes and Pt/C
cathodes, at 80 °C, of: (i) Capillary-fed cell with PTFE at the anode,
(ii) Capillary-fed cell without PTFE at the anode, (iii) control,
conventional, bubbled cell with Zirfon PERL UTP 500 as inter-electrode
separator, (iv) control, conventional, bubbled cell with polyether
sulfone filter (8 μm average pore diameter) as inter-electrode
separator, and (v) control, conventional, bubbled cell with polyether
sulfone filter (8 μm average pore diameter) as inter-electrode
separator, where the anode is welded to its bipolar plate. c
Comparison of capillary-fed electrolysis cell (85 °C) with commercial
alkaline and PEM electrolysis cells (90 °C) representative of the
state-of -the-art. Polarisation curves of: (i) the capillary-fed
electrolysis cell with PTFE at the anode at 85 °C, (ii) commercial
alkaline electrolysis cell at 90 °C 24 representative of
the state-of-the-art, and (iii) commercial PEM electrolysis cell at
90 °C20
representative of the state-of-the-art. d Performance of
capillary-fed electrolysis cell at a fixed 100% energy efficiency (HHV)
(85 °C). (i) Current density at 85 °C of the capillary-fed
electrolysis cell in Fig. 3c(i)
when poised at a fixed cell voltage of 1.47 V, which equates to 100%
energy efficiency (HHV). Supplementary Fig. 10
provides data from 1-day and 30-day tests at 80 °C and room
temperature, with a large reservoir that was regularly manually
replenished.
In commercial systems, bipolar plates are
normally employed to carry current into and out of electrolysis cells.
Within a cell, each bipolar plate connects electrically to their
corresponding electrode via a conducting gas diffusion layer (also
called a porous transport layer in PEM electrolysis cells) (Fig. 3a).
The present work sought to replicate this arrangement to make fair
comparisons with commercial cells. This was done by combining the
bipolar plate and conducting gas diffusion layer into a thick sheet of
Ni with many small holes to allow evolved gases to exit (Supplementary
Fig. 6).
This bipolar plate/flow field structure was housed within a gas
chamber that could be sealed to the external environment. Leak-tight
bolts through the walls of the gas chambers were used to press the
bipolar plates and their attached electrodes against the polyether
sulfone separator (Supplementary Fig. 6).
The gas chambers were flushed with nitrogen prior to operation.
For the anode, a fine Ni mesh was
electrocoated with a NiFeOOH electrocatalyst, as previously described
by Benedetti and colleagues23
and then spot-welded to its bipolar plate, without loss of catalyst,
using the approach depicted in Supplementary Fig. 7.
No carbon was present in the anode to avoid oxidative carbon corrosion
currents, which can lead to highly misleading results.
In some cells, a 60 wt% dispersion of
polytetrafluoro-ethylene (PTFE, also known as Teflon) was
included in the electrocoating solution. Catalyst coatings from the
resulting solution were found to display enhanced anode performance.
For the cathode, a Pt/C electrocatalyst
was deposited as previously described by Liu et al.24,
to a loading of 0.5 mg cm−2 Pt on a conducting, carbon
paper gas diffusion layer. As the carbon paper could not be welded,
the cathode was pressed tightly against its bipolar plate as described
in Supplementary Fig. 6.
The structure of the cathode is described in detail in Supplementary
Note
3.
Two-electrode measurements, including the
polarisation curves, were recorded between the anodic and cathodic
bipolar plates, across the cell, as they would be in a commercial
cell. The electrolyte was 27 wt% aqueous KOH.
The current densities reported here are
relative to the geometric area of the electrodes that were covered
with electrocatalysts. Prior to testing, the current produced by the
metal structures in the cell that were not coated with catalyst, was
determined by operating the cell without catalysts, with and without
the gas chambers filled with air. Currents of <0.025 A cm−2
were observed up to a cell voltage of 2.1 V.
With catalysts, and with the gas chambers
initially filled with air, oxygen depolarisation of the cathode
occurred only up to 0.030 A cm−2, whereafter hydrogen
production overwhelmed oxygen ingress (Supplementary Fig. 8).
The CFE cell with the above electrodes
and polyether sulfone separator was then tested at 80 °C, giving the
current-voltage curves in Fig. 3b(i)
(with PTFE in the anode) and Fig. 3b(ii)
(without PTFE in the anode) (Source DataFigs. 24). As can be seen in
Fig. 3b(i),
with PTFE incorporated in the anode, the cell required a voltage of
1.59 V to drive water electrolysis at 1 A cm−2. Without
PTFE in the anode, 1.61 V was needed at 1 A cm−2 (Fig. 3b(ii)).
For comparative purposes, and to
investigate changes in the cell resistance, identical zero-gap water
electrolysis cells that were fully flooded with liquid electrolyte,
causing them to produce gases in the form of bubbles, were also
prepared. These control cells employed the same cathode and anode
(without PTFE) as above, with 27 wt% KOH. They were tested at the same
temperature of 80 °C.
With the well-known commercial alkaline
separator, Zirfon PERL UTP 500, the resulting, bubbled control cell
required 1.86 V to produce 1 A cm−2 (Fig. 3b(iii)).
When the Zirfon was replaced with the
polyether sulfone separator, the cell needed 1.74 V at 1 A cm−2
(Fig. 3b(iv)),
which was 0.12 V lower. This equates to a decrease in the cell
resistance of 120 mΩ cm2, which can be almost entirely
attributed to the lower ionic resistance of the polyether sulfone
separator compared to Zirfon PERL UTP 500 at 80 °C (Supplementary
Fig. 5).
Following operation, a contact resistance
was found to have developed between the anode and its bipolar plate.
This contact resistance was due to the formation of a layer of poorly
conducting Ni oxide on the contacting Ni surfaces by the oxygen
produced at the anode.
To overcome this contact resistance, the
control cell with the polyether sulfone separator, was modified by
welding its anode to its corresponding bipolar plate. The anodes of
the capillary-fed cells had been welded to their bipolar plates for
the same reason. The effect was to decrease the voltage required at
1 A cm−2 to 1.66 V (Fig. 3b(v)),
which equates to a further decline of 0.08 V and an 80 mΩ cm2
lower cell resistance. This result is relevant insofar as some
commercial water electrolysis cells still employ compression to create
electrical contact2,
rather than welding, which is routinely used in other types of
electrolysis cells25.
The contact resistance between the
cathode and its bipolar plate after operation was separately measured
to be a much lower 35 mΩ cm2. The reducing environment of
the cathode avoids formation of poorly conducting surface layers.
Accordingly, the capillary-fed cell in
Fig. 3b(i)
significantly outperformed its conventional, bubbled, control cells
employing the same electrodes and electrolyte in an identical zero-gap
configuration.
The performance of the above CFE cell
with PTFE at the anode, was also compared with data from commercial
alkaline and PEM electrolysis cells representative of the current
state-of-the-art20,26.
As the commercial data had been collected at a higher temperature of
90 °C, the CFE cell was tested at 85 °C.
As can be seen in Fig. 3c,
the CFE cell substantially outperformed the commercial cells. It
improved on alkaline cells, which are typically operated commercially2
at ~0.20.8 A cm−2. It also improved on commercial PEM
cells, which must be operated at higher current densities in the
1.53.0 A cm−2 range to be commercially viable20.
To produce typical commercial operating
current densities of 0.5 A cm−2 for alkaline and 1.8 A cm−2
for PEM, the commercial cells required ~1.77 V (Fig. 3c(ii)(iii)),
equating to a cell energy efficiency of ~83% (HHV) and an energy
consumption of ~47.5 kWh kg−1 H2. By contrast,
the alkaline CFE cell required only 1.506 V at 0.5 A cm−2
(Fig. 3c(i)),
which represents a cell energy efficiency of 98% (HHV) with
consumption of only 40.4 kWh kg−1 H2, or
3.64 kWh Nm−3 H2 (Table 2).
The ~7.1 kWh kg−1 decrease in energy consumption surpasses
the IRENA 2050 target2
of <42 kWh kg−1 and realises a 15% improvement in cell
energy efficiency.
Table 2 Performance at 85 °C of the
capillary-fed electrolysis cell with PTFE at the anode.
When held at the thermoneutral voltage
for water electrolysis, 1.47 V at 85 °C, which equates to 100% energy
efficiency (HHV), the capillary-fed cell produced a constant ~0.3 A cm−2
(Fig. 3d).
As far as the authors are aware, no water electrolysis cell, either
alkaline or PEM, has ever produced 0.3 A cm−2 at 100%
energy efficiency (HHV).
The CFE cell also demonstrated sustained
stable performance over extended periods from 1 working day to 30 days
continuously at 80 °C and room temperature, respectively, with
periodic replenishment of the consumed water to the reservoir
(Supplementary Fig. 10).
Water spontaneously migrated from the reservoir up the separator,
possibly under an osmotic as well as a capillary impulse to counteract
increases in the KOH concentration in the separator due to water
consumption at the electrodes. No KOH build up or crystallisation was
observed in or on the separator.
The origin of the high performance of the
capillary-fed electrolysis cell
The unprecedented performance of the CFE
cell in Fig. 3b(i)
and Fig. 3c(i)
may be explained in part by the well-known high activity of the
NiFeOOH anode electrocatalyst25,27,28,29,30
and Pt/C cathode electrocatalyst, and the low ionic resistance of the
polyether sulfone separator (Table 1).
Galvanostatic electrochemical impedance
spectroscopy could be used to determine the resistances in the
capillary-fed cell under active water electrolysis, but not their
origin. For example, at 0.35 A cm−2 and 80 °C, the cell
demonstrated a series resistance of ~40 mΩ cm2 that was
mainly but not completely attributable to the low ionic resistance of
the polyether sulfone separator (Supplementary Fig. 11).
To elucidate the major elements that
contributed to the high performance of the CFE cell at 80 °C, the
control, conventional, bubbled cell with Zirfon PERL UTP 500 separator
(Fig. 3b(iii))
was therefore taken as a baseline. That cell needed 1.86 V to generate
1 A cm−2 (Fig. 3b(iii)).
When the Zirfon was replaced with the
polyether sulfone separator, the voltage required at 1 A cm−2
was 1.74 V (Fig. 3b(iv)),
which was 0.12 V less. As noted, the resulting 120 mΩ cm2
reduction in resistance is almost all due to the lower ionic
resistance of the polyether sulfone separator.
The effect of welding the anode of the
bubbled cell with the polyether sulfone separator, to its bipolar
plate was to further decrease the voltage needed at 1 A cm−2
to 1.66 V (Fig. 3b(v)).
The resulting 80 mΩ cm2 decrease in resistance arose by
elimination of a contact resistance that developed between the anode
and its bipolar plate during operation.
The CFE cell achieved still lower
voltages. Without PTFE at the anode, it required a cell voltage of
1.62 V at 1 A cm−2 (Fig. 3b(ii)),
which is 0.04 V lower, equating to a further 40 mΩ cm2
decrease in resistance.
With PTFE at the anode, the capillary-fed
cell required only 1.59 V at 1 A cm−2 (Fig. 3b(i)),
which constituted a still further decrease of 0.03 V, with an
additional lowering in cell resistance of 30 mΩ cm2.
These improvements may be attributed to
the combined contribution of several possible factors, including the
following.
Firstly, the CFE cells were largely
bubble-free during operation. This was confirmed by comparing voltage
fluctuations due to bubble formation and release at a series of fixed
current densities12.
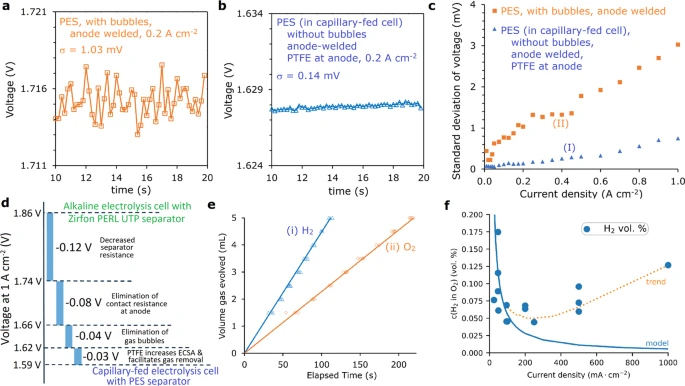
Fig. 4a
and b show the results and analysis of steady-state
chrono-potentiometric measurements of the bubbled control cell in
Fig. 3b(v)
and the capillary-fed cell in Fig. 3b(i),
respectively, over the last 10 s of a 20 s period at 0.2 A cm−2.
The voltage of the bubbled cell (Fig. 4a)
(Source DataFigs. 24) was characterised by a noisy response
attributable to the nucleation, growth, coalescence, and release of
gas bubbles. The standard deviation (σ) of the voltage signal
for the bubbled cell was 1.03 mV. By contrast, the voltage of the
capillary-fed cell under the same conditions was remarkably steady
(Fig. 4b),
with a significantly lower σ of 0.14 mV. The steady voltage
signal and low σ value of the capillary-fed cell is consistent
with bubble-free operation.
(At room
temperature with 27 wt% KOH unless stated otherwise). a Voltage
profile due to bubble formation. Voltage fluctuations in the bubbled,
control cell in Fig. 3b(v)
over the last 10 s of a 20 s step at 0.2 A cm−2
(σ = standard deviation). b Voltage profile in the
capillary-fed cell. Voltage fluctuations in the cell in Fig. 3b(i)
over the last 10 s of a 20 s step at a fixed 0.2 A cm−2.
c Voltage profile uniformity as a function of current density.
Plot of the standard deviation in voltage for the cells in: (I) Fig. 3b(i),
and (II) Fig. 3b(v),
at different current densities, each held for 20 s, with the standard
deviation measured over the last 10 s of the 20 s period. The bubbled
control cell in Fig. 3b(v)
displays much larger fluctuations in voltage due to bubble formation
than the capillary-fed cell in Fig. 3b(i),
which exhibits largely bubble-free operation at ≤0.2 A cm−2,
and substantially bubble-free operation at 0.251 A cm−2.
d Voltage declines and their origins. Waterfall plot showing
the voltages declines observed at 1 A cm−2 (80 °C) and
their sources. e Faradaic efficiency. Rates of: (i) hydrogen
generation, and (ii) oxygen generation by the capillary-fed
electrolysis cell in Fig. 3b(i),
at a fixed 0.350 A/cm2 at atmospheric pressure, after
30 min. The data points indicate the measured volumes. The solid lines
plot the theoretical rate of gas generation at 100% Faradaic
efficiency. The overall Faradaic efficiency of the cell, determined by
comparing the slopes of the measured and theoretical data, including
both gases, was 99.5 ± 1.3%. f Hydrogen crossover. The data
points show the hydrogen crossover into the anodic oxygen stream of
the capillary-fed electrolysis cell in Fig. 3b(i)
as a function of current density, at room temperature and atmospheric
pressure. Each data point was collected after operating the cell for
30 min at the relevant current density. The solid line shows the gas
crossover expected from diffusion only31,
32. The dashed line depicts the trend in the data.
Steady-state chrono-potentiometric
measurements of bubbled and capillary-fed cells over a range of
current densities from 0.01 to 1 A cm−2 and analysis of the
results permitted the construction of the standard deviation vs.
current density plot in Fig. 4c.
The bubbled cell demonstrated a relatively steep increase in σ
values commencing from the lowest current density of 0.01 A cm−2.
By contrast, the capillary-fed cell had much lower and flatter σ
values between 0.01 and 0.2 A cm−2, before the σ
values increased modestly as the current density was raised to 1 A cm−2.
The results suggest that the capillary-fed cell was largely
bubble-free up to and including 0.2 A cm−2, and
substantially bubble-free between 0.25 and 1 A cm−2. At
1 A cm−2, the capillary-fed cell displayed a σ value
of 0.75 mV, which was comparable to the bubbled cell at ~0.09 A cm−2,
suggesting that <10% of the current went into gas production via
bubble formation (Supplementary Fig. 9).
These findings are supported by the fact
that the performance of the capillary-fed cell in Fig. 3c(i)
mainly differed from the commercial PEM cell in Fig. 3c(iii)
in having a lower onset potential; the slopes of the curves were
similar. Previous studies have demonstrated that bubble-free operation
decreases the onset potential10,11,13.
The bubble-producing cells in Fig. 3b(iii)(v)
had onset potentials of ≥1.45 V, while that of the capillary-fed cell
in Fig. 3b(i)
was ~1.39 V.
Bubble-free gas evolution likely
contributed to the voltage decline insofar as the electrodes were not
masked with bubbles, leaving the catalytic sites on the electrodes
more available for reaction. This probably enhanced the overall
performance of, particularly, the most active catalytic sites that are
the source of most bubbles and the first to be blocked by bubbles. The
resulting fuller use of the available electrocatalytic sites likely
improved performance.
The avoidance of bubble formation at
≤0.2 A cm−2, which was likely due to the gas-liquid
interface being within diffusion distance of the electrode, may also
have decreased the supersaturation of the electrolyte, leading to a
voltage decline10,13.
According to the Nernst equation, elevated gas concentrations increase
E°33.
At higher current densities, supersaturation may have been needed at
some electrode locations to produce the few bubbles observed.
The architecture of the capillary-fed
cell may also have contributed insofar as it ensured that the flow of
water toward each electrode did not counter the flow of gas away from
the electrode. That is, the architecture of the capillary-fed cell
inherently avoided the counter multiphase flows present in
conventional, bubbled water electrolysers.
At this stage it is not possible to
determine the absolute contribution of each of the above factors, but
cumulatively they resulted in a 40 mΩ cm2 lower resistance
in the capillary-fed cell without PTFE at the anode (Fig. 4d).
With PTFE at the anode, additional
improvements were realised in the capillary-fed cell (Fig. 3b(i)).
Electron micrographs indicated that the PTFE was dispersed as
needle-like structures on the anode (Supplementary Fig. 12).
The double layer capacitance of the anode increased by ~12-fold, from
0.46 mF cm−2 without PTFE to 5.50 mF cm−2 with
PTFE (Supplementary Fig. 13).
This suggests that the electrochemically/catalytically active surface
area (ECSA) of the anode increased significantly. As the specific
capacitance of the catalyst with PTFE is not known, the precise scale
of the increase could not be determined. However, the PTFE clearly
increased the porosity of the electrocatalytic layer and this
increased the ECSA.
A body of previous work has also
demonstrated that PTFE surfaces on an electrocatalyst may scavenge,
coalesce and transport away newly formed, dissolved gases34.
PTFE is highly aerophilic, with low surface energy. A similar
mechanism may have been partly responsible for the improved,
bubble-free performance of the anode when PTFE was incorporated. That
is, the PTFE on the anode may have amplified the catalytic performance
by facilitating migration of newly formed gas along its aerophilic
surfaces, across the gas-liquid interface, to thereby avoid gas bubble
formation. As described in Supplementary Note
3, it is potentially significant that the same elements of
aerophilic PTFE surface pathways for gas transport across the
gas-liquid interface were also present on the cathode, which exhibited
similarly bubble-free performance.
Whatever the origin of the improved
performance, the incorporation of PTFE in the anode contributed a
decrease in voltage of 0.03 V at 1 A cm−2, equating to a
decline in resistance of 30 mΩ cm2 (Fig. 4d).
In summary, the capillary-fed cell
continues the evolution of water electrolysis cells by systematic
decreases in cell resistance, as illustrated in Fig. 1.
However, the large net decrease realised did not have a single origin.
It involved many smaller contributions that, cumulatively, led to a
270 mΩ cm2 reduction in cell resistance at 80 °C, over the
standard commercial configuration of a bubbled, zero-gap cell with a
Zirfon PERL UTP 500 separator (Fig. 4d).
Faradaic efficiency approaching 100%, and
low hydrogen crossover
The Faradaic efficiency and gas crossover
are important features of electrolysis cells. The latter is a
potential safety issue as a hydrogen stream containing >4.6% oxygen,
or an oxygen stream with >3.8% hydrogen, constitutes an explosive
mixture (at 80 °C)10.
The volumes of hydrogen and oxygen
produced by the best performing capillary-fed cell at a fixed
0.35 A cm−2 were measured (Fig. 4e).
There was a close agreement between the measured volumes and what
would be expected if all electrons went into water electrolysis,
giving an overall Faradaic efficiency, including both gases, of
99.5 ± 1.3% (Source DataFigs. 24).
To measure the extent of hydrogen gas
crossover, the CFE cell with PTFE at the anode was connected to a gas
chromatograph and measurements were taken of the hydrogen impurity in
the anodic oxygen stream. Figure 4f
shows the concentration of hydrogen in the anodic oxygen stream as a
function of current density. These results, which fell between 0.04
and 0.14 vol% at 0.11.0 A cm−2, are notably lower than
reported rates of hydrogen crossover with conventional separators
(Supplementary Table 5).
The oxygen impurity in the cathodic
hydrogen stream was similarly examined, however no crossover could be
observed within the detection limit of 0.001 vol% (10 ppm).
The low crossover of the capillary-fed
cell may be ascribed to a different and unique mechanism of gas
crossover.
In fully flooded, bubbled alkaline
electrolysis cells, gas crossover is known to occur in two ways34,35:
(1) diffusion of dissolved gas through the liquid in the separator,
and (2) advective flow of liquid electrolyte, carrying dissolved gas
and bubbles with it, through the porous separators that are generally
used. The latter is driven by fluctuating or perpetual pressure
differentials across the separator, and is, by far, the larger
contributor, producing orders of magnitude more crossover35,36.
To decrease the advective cross flow of electrolyte, separators in
alkaline electrolysis are designed to have the smallest possible pores
(<0.15 μm diameter)35.
The rate of diffusion-based crossover in alkaline cells is exceedingly
low because the high levels of K+ and OH− ions
in alkaline electrolytes salt-out dissolved gases like hydrogen and
oxygen, which have very low solubilities and diffusion coefficients in
alkaline electrolytes31,32.
In PEM electrolysis cells, the proton
exchange membranes are non-porous. This eliminates advective flows as
a significant mechanism of gas crossover since the de-ionised water
used in such cells is unable to freely pass through the membrane36,37.
The only available mechanism of gas crossover is diffusion3.
The combined solubility and diffusion coefficients of hydrogen and
oxygen are, however, 40120-times higher in de-ionised water than in
typical alkaline electrolytes at 80 °C31,32,36.
While advective crossover is absent in PEM cells, diffusion-based
crossover, with balanced hydrogen and oxygen pressures on opposite
sides of the PEM membrane, is therefore usually notably larger than in
alkaline systems (Supplementary Table 5)36,37,38.
The industry has overcome this issue by using very high hydrogen
pressures but only atmospheric oxygen pressures on the opposite sides
of the PEM membrane. This maintains the produced hydrogen free of
oxygen impurity37.
The CFE cells are in the unique position
of avoiding advective crossover, whilst also having low
diffusion-based crossover because of the high molarity alkaline
electrolyte used.
Advective crossover is not available as a
mechanism of crossover in capillary-fed cells since there are no
unrestricted bodies of liquid electrolyte on both sides of the
separator that are free to flow through it under the influence of a
pressure differential. That is, only diffusion-based crossover is
possible. However, a high molarity alkaline electrolyte is used, and
this provides for only a small diffusion-based crossover.
This explanation is supported by the fact
that, whereas separators in conventional, fully flooded, bubbled
alkaline electrolysis cells can only minimise crossover by having very
small pores (<0.15 μm), the capillary-fed cell has the largest of the
available pores (8 μm) but still exhibits minor gas crossover.
It is further supported by the
observation in Fig. 4b,
that, at current densities of ≤0.2 A cm−2, the measured
rates of gas crossover lie within the range expected for diffusion
only, which is depicted as the solid line. At current densities above
0.2 A cm−2 however, hydrogen crossover diverges from the
diffusion-only model and trends moderately upward (Fig. 4f,
orange dotted line). This is consistent with the observed minor bubble
formation above 0.2 A cm−2 in Fig. 4c,
which may involve supersaturation at a few locations on the
hydrogen-generating cathode36.
Such localised supersaturation would create a partial pressure
gradient resulting in moderately increased hydrogen crossover.
Simplification of the balance-of-plant
Capillary-fed electrolysis (CFE) cells
may be readily incorporated into a bipolar cell stack of the type used
in commercial electrolysers (Supplementary Fig. 2).
The engineering system required to manage such a stack, known as the
balance-of-plant, may then be compared with a typical, conventional
balance-of-plant13.
Supplementary Fig. 3
depicts this comparison. As can be seen, the balance-of-plant needed
for the CFE cell concept (Supplementary Fig. 3b)
is notably less complex than that needed for a conventional, bubbled
electrolysis cell (Supplementary Fig. 3a).
There is, firstly, no need for pumped
liquid circulation, or for gas/liquid separators, as there are no
liquid-enveloped gas bubbles that need to be constantly swept away
from the electrodes (Supplementary Fig. 3b).
The gas-liquid froths that are generated when gas bubbles are produced
must normally be pumped to separator tanks for partitioning into bulk
liquid and gas phases (Supplementary Fig. 3a).
The high-volume electrolyte pumps
(depicted below each separator tank in Supplementary Fig. 3a)
are therefore also not needed, nor are their associated piping and
fittings, namely, the anolyte and catholyte forward and return lines.
Pumps and piping of this type are expensive because they need to avoid
corrosion by KOH (in the case of an alkaline electrolyser) or leaching
of metal ions into the de-ionised water used (in the case of a PEM
electrolyser) and comply with stringent hydrogen or oxygen safety
standards.
In a conventional balance-of-plant, de-ionised
make-up water would typically be added to each electrolyte circulation
loop, via the scrubbers, from a pressurised dispensing system (as
shown at the top of Supplementary Fig. 3a).
In the capillary-fed balance-of-plant, a comparable pressurised
dispensing system would be needed to add de-ionised water to the
individual reservoirs (as shown at the bottom of Supplementary Fig. 3b).
Such arrangements are already used in the chlor-alkali industry, where
make-up water and brine are routinely dispensed to individual half
cells39.
Another feature of conventional
balance-of-plants are the need for water-cooled chillers to remove the
excess heat produced during electrolysis (Supplementary Fig. 3a).
The high energy efficiency of the capillary-fed cell produces only
modest Joule heating during operation however, avoiding the need for a
water-cooled chiller as demonstrated in Supplementary Note
1, Supplementary Tables 12,
and Supplementary Data 1.
Instead, air-cooling or radiative self-cooling of the stack may be
possible.
Because of the need for large volumes of
liquid to remove and separate the gas bubbles formed, conventional
commercial cell stacks and their balance-of-plants typically contain
~10,000 L of water per MW. By contrast, as shown in Supplementary Note
2 and Supplementary Table 3,
a, the CFE system is likely to require only ~500 L of water per
MW.
One effect of a lower water requirement
is a reduced need for water purification and replacement in the
balance-of-plant. This is especially relevant to PEM electrolysers,
which require on-going de-ionisation of the water content, using a
costly class 1 de-ioniser, to achieve a stack lifetime of ~70,000 h.
In alkaline electrolysers, only the input make-up water needs
deionisation, with the entire body of liquid electrolyte typically
needing replacement every 56 years8.
A final advantage of the capillary-fed
cell system is its capacity to avoid the wasteful and corrosive high
voltage shunt currents13
that flow between cells along the catholyte and anolyte return lines
in conventional alkaline electrolysers. In the capillary-fed
balance-of-plant, the only potential liquid conduction path between
cells is along the water dispensing line shown at the bottom of
Supplementary Fig. 3b.
But this contains de-ionised water that is not conductive.
These simplifications in the
balance-of-plant lead to downward pressure on electrolyser CAPEX. They
may also lower the power consumption of the balance-of-plant, further
decreasing the energy needed per kg hydrogen.
Discussion
This work introduced the CFE cell
concept. Using existing catalysts, with Faradaic efficiencies
approaching 100%, and low hydrogen crossover, this architecture
significantly improved the energy efficiency of the water
electrolysis cell. At the operating current density used in many
commercial alkaline electrolysis cells of 0.5 A cm−2, a
cell voltage of only 1.506 V was needed to produce hydrogen at
85 °C. This represents a cell energy efficiency of 98% (HHV), with
consumption of just 40.4 kWh kg−1 H2, or
3.64 kWh Nm−3 H2. This result surpasses
commercial electrolysis cells, which consume ~47.5 kWh kg−1 H2,
and exceeds the 2050 IRENA target2
of <42 kWh kg−1.
The
CFE cell also allows for a notably simplified balance-of-plant,
further reducing energy consumption and putting downward pressure
on CAPEX.
These substantial improvements on
present-day state-of-the-art electrolysis cells translate to
direct declines, of similar proportion, in the levelised cost of
hydrogen. Combined with the promise of a simplified system
balance-of-plant, they bring cost-competitive renewable hydrogen
closer to reality.
Methods
Materials
Porous polyether sulfone (PES) filters of
0.03 μm, 0.45 μm, 1.2 μm, 5 μm, and 8 μm average pore diameters (Pall
Corporation and Sterlitech), Carbon black (AkzoNobel), 10% Pt on
Vulcan XC-72 (Premetek), NiCl2.6H2O and FeCl2.4H2O
(Sigma-Aldrich), PTFE (60 wt.% dispersion in alcohols/H2O;
Sigma-Aldrich, 510211), Nafion dispersion (5% in in
alcohols/water; Sigma-Aldrich), SigracetTM 22BB carbon
paper (Fuel Cell Store), Zirfon PERL UTP 500 (Agfa), KOH 90%, flakes
(Sigma-Aldrich), Ni mesh (Century Woven, Beijing).
Porosity and in-plane flow model values
Porosity was determined by comparing the
weight of dry and saturated polyether sulfone. Cross-sectional area
was calculated by multiplying width (1 cm, nominal) by thickness (140 μm,
measured). Average pore diameter was determined using capillary flow
porometry. Contact angle was measured using the captive bubble
technique. Viscosity16
and surface tension17
were obtained from the literature.
In-plane flow rate measurements
In-plane flow rates were measured using
the setup shown in Supplementary Fig. 4(a).
For each experiment a 1 cm-wide polyether sulfone microfiltration
strip was encased in plastic sheathing of length L to avoid
electrolyte evaporation; 1.5 mm at each end of the polyether sulfone
strip was left exposed. To the top of the strip was clamped a pad
composed of several layers of absorbent paper. The bottom of the strip
was dipped in electrolyte. The weight of the assembly was measured
over time. An example weight vs. time plot is provided in
Supplementary Fig. 4(b)
and shows an initial curved response corresponding to initial filling
of the strip (Washburn flow), followed by a linear response
corresponding to continuous flow through the fully wetted strip (Darcy
flow). The in-plane flow rate was taken as the slope of the linear
part of the weight vs. time plot.
Ionic resistance of polyether sulfone
separator
The ionic resistance of the polyether
sulfone separator was determined using a method described in the
literature40
and the setup shown in Supplementary Fig. 5.
The conductivity of the KOH electrolyte was measured with and without
a polyether sulfone separator present using a four-point conductivity
probe (Mettler Toledo Sevencompact with Inlab ISM-731 Probe) as
described in Supplementary Fig. 5.
Electrode preparation
The hydrogen-evolving cathode was prepared using a literature method24.
100 mg 10% Pt on Vulcan XC-72, 0.8 mL 5 wt% Nafion solution, 1.5 mL
deionised water, and 3 mL iso-propanol were combined and sheared at
10,000 rpm for 5 min using a homogeniser (IKA T25). The resulting
dispersion was air brushed onto the microporous layer side of a
1 cm × 1 cm carbon fibre paper (Sigracet 22BB) to a loading of
0.5 mg cm−2 Pt.
The oxygen-evolving anode was prepared
following a literature method23.
Prior to use, Ni mesh (200 LPI, ų 50 μm wire, 75 μm aperture) was
cleaned by ultrasonication in isopropyl alcohol for 10 min and dried,
then pickled in 5 M HCl for 10 min, rinsed with deionised water, and
dried. After taping as shown in Supplementary Fig. 7,
electrodeposition was performed at room temperature in a 3-electrode
cell comprising 1 cm × 1 cm Ni mesh working electrode, oversized Ni
mesh counter electrode, and Ag/AgCl (3 M NaCl) reference electrode.
The nickel mesh was placed in an electrocoating solution that
comprised of a 3:1 mixture of NiCl2.6H2O
(0.075 M) and FeCl2.4H2O (0.025 M) (following
Fig. 8(c) and Fig. 1(a) in ref.
23), along with 1 M KCl supporting electrolyte (following
Fig. 8(b) in ref.
23). The nickel mesh immersed in the electrocoating solution
was coated with NiFe by repeated cycling using cyclic voltammetry
between −1.0 V and −0.2 V (vs. Ag/AgCl) at 10 mV s−1 until
a charge of 17 C had been deposited (following Fig. 1 in ref.
23). A BioLogic VSP potentiostat was used. The lower voltage
of −1.0 V allowed for inclusion of a PTFE dispersion, without
precipitation; the upper voltage of −0.2 V was found to provide the
best catalytic performance. A dispersion of PTFE could be incorporated
into the anode by including the equivalent of 10 g L−1 PTFE
in the electrocoating solution. Following electrodeposition, the
working electrode was rinsed with deionised water and dried at room
temperature. The electrocoating tape was then removed and the anode
was welded to the bipolar plate as shown in Supplementary Fig. 7.
Capillary-fed cell
A picture of the capillary-fed cell used
experimentally is provided in Supplementary Fig. 6.
The polyether sulfone separator (8 μm average pore diameter) was
initially soaked in deionised water and then kept in 27 wt% KOH
electrolyte overnight. Ni gas diffusion bipolar plates measured
1.4 cm × 1.4 cm × 0.1 cm and included numerous 1 mm holes to allow
evolved gases to exit the electrode. They performed the function of
both the bipolar plate and the gas diffusion layer/porous transport
layer in electrolysis cells. Bare Ni mesh was spot-welded to the Ni
gas diffusion-bipolar plate on the cathode side and then pressed
tightly against the rear of the Sigracet cathode substrate. The
contact resistance was 35 mΩ cm2. The Ni mesh anode was
spot-welded to the Ni gas diffusion bipolar plate on the anode after
coating with NiFe catalyst as described in Supplementary Fig. 7.
The capillary-fed cell had the architecture: Ni bipolar plate/Ni
mesh/Pt-C on carbon paper/ polyether sulfone separator/NiFe-coated Ni
mesh/Ni bipolar plate. The anode and cathode were pressed against the
polyether sulfone separator by leak-tight bolts passing through the
gas chambers (Supplementary Fig. 6).
Finally, the reservoir into which the bottom end of the polyether
sulfone separator was dipped, was filled with 27 wt% KOH electrolyte.
Electrochemical measurements
Electrochemical measurements were
performed using a BioLogic VMP3 potentiostat. Linear sweep voltammetry
was performed by sweeping the cell voltage upward from 1.21.4 V at
10 mV s−1 until the current density reached 1.5 A cm−2.
Measurements at 80 °C or 85 °C were performed upon temperature
equilibration after placing the capillary-fed cell into an oven at
that temperature. Galvanostatic electrochemical impedance spectroscopy
(GEIS) was performed at 0.350 A cm−2 DC bias, 0.050 A cm−2
AC perturbation, and between 100 kHz and 100 mHz.
Faradaic efficiency
Faradaic efficiency was calculated by
comparing the measured volumes of hydrogen and oxygen produced at the
cathode and anode, of a capillary-fed cell, respectively, at a fixed
current of 0.350 A cm−2 at room temperature, with the
expected volumes of hydrogen and oxygen if all electrons resulted in
gas evolution (including the expected water vapour content of the
gases and considering the gas crossover, which was measured at the
same time). Gas volumes were measured by collecting the produced gas
in a submerged, upturned burette. The cell was operated for 30 min at
0.350 A cm−2 prior to taking each set of measurements.
Hydrogen crossover
The concentration of hydrogen in the
anode oxygen stream of a capillary-fed cell operated at different
current densities at room temperature was determined by piping the
anode gas output for analysis to a gas chromatograph (Shimadzu GC8A),
operating with thermal conductivity detection and flame ionisation
detector and argon as a gas carrier. The hydrogen and oxygen peaks
were identified by their retention times and integrated to determine
the relative quantities of gas present.
The Future of Hydrogen.
(International Energy Agency, Paris, France, 2019).
Green Hydrogen Cost Reduction:
Scaling up Electrolysers to Meet the 1.5 °C Climate Goal.
(International Renewable Energy Agency, Abu Dhabi, 2020).
OPEX is, by far, the major component of the LCOH for the
following reason: Fig. ES1 in this reference provides an
average electrolyser CAPEX (today) of USD$770 per 1 kW
capacity. Over 10 years of continuous operation the
electricity consumed by that 1 kW capacity, which will
comprise most of the OPEX, will be:
24 × 365 × 10 = 1,752,000 kWh. At the average electricity
price of USD$53/MWh, this gives a total OPEX of at least
USD$91,104, which is 118-times larger than the CAPEX. The
dominance of the OPEX over the CAPEX in the LCOH is
projected to increase. Using the best future CAPEX of
USD$130/kW and electricity price of USD$20/MWh over 10 years
of continuous operation: the OPEX of USD$35,040 will be
270-times larger than the CAPEX. The IRENA 2050 cell energy
consumption target is given in Table 6 (Stack electrical
efficiency).
van Renssen, S. The hydrogen
solution? Nat. Clim. Change10, 799801
(2020).
Maric, R. & Yu, H. in
Nanostructures in Energy Generation, Transmission and
Storage (ed Yanina Fedorenko) 95117 (IntechOpen, 2019).
Lund, P. D., Lindgren, J., Mikkola,
J. & Salpakari, J. Review of energy system flexibility
measures to enable high levels of variable renewable
electricity. Renew. Sustain. Energy Rev.45,
785807 (2015).
Bogdanov, D. & Breyer, C. North-East Asian
Super Grid for 100% renewable energy supply: Optimal mix of energy
technologies for electricity, gas and heat supply options. Energy
Convers. Manag.112, 176190 (2016).
Godula-Jopek, A. Hydrogen Production by
Electrolysis, Wiley-VCH, 2015, and references therein. ISBN:
978-3-527-33342-4.
Winther-Jensen, O., Chatjaroenporn, K.,
Winther-Jensen, B. & MacFarlane, D. R. Towards hydrogen production
using a breathable electrode structure to directly separate gases in
the water splitting reaction. Int. J. Hydrog. Energy37,
81858189 (2012).
Tiwari, P., Tsekouras, G., Wagner, K.,
Swiegers, G. F. & Wallace, G. G. A new class of bubble-free
water electrolyzer that is intrinsically highly efficient. Int. J.
Hydrog. Energy44, 2356823579 (2019).
Tsekouras, G. et al. Insights into the
phenomenon of bubble-free electrocatalytic oxygen evolution
from water. Sustain. Energy Fuels5, 808819, and
references therein,
https://doi.org/10.1039/D0SE01633K (2021).
Swiegers, G. F. et al. The prospects of
developing a highly energy-efficient water electrolyser by eliminating
or mitigating bubble effects. Sustain. Energy Fuels5,
12801310 (2021). and references therein.
Iversen, S. B., Bhatia, V. K., Dam-Johansen,
K. & Jonsson, G. Characterization of microporous membranes for use in
membrane contactors. J. Membr. Sci.130, 205217 (1997).
Properties of Aqueous Solutions of
Electrolytes. (CRC Press, 1992).
Wang, P., Anderko, A. & Young, R. D.
Modeling Surface Tension of Concentrated and Mixed-Solvent Electrolyte
Systems. Ind. Eng. Chem. Res.50, 40864098 (2011).
Technical Data Sheet, ZIRFON PERL UTP 500,
Separator membrane for alkaline electrolysis. (Agfa, 2020).
Schalenbach, M., Lueke, W., Lehnert, W. &
Stolten, D. The influence of water channel geometry and proton
mobility on the conductivity of Nafion®. Electrochim. Acta
214, 362369 (2016).
Debe, M. K. et al. Initial Performance and
Durability of Ultra-Low Loaded NSTF Electrodes for PEM Electrolyzers.
J. Electrochem. Soc.159, K165K176 (2012).
Kim, J.-H. et al. Low-cost and energy
efficient asymmetric nickel electrode for alkaline water electrolysis.
Int. J. Hydrog. Energy40, 1072010725 (2015).
Schalenbach, M., Kasain, O. & Mayrhoffer, K.
J. J. An alkaline water electrolyzer with nickel electrodes enables
high current density operation. Int. J. Hydrog. Energy43,
11931198 (2018).
Sakita, A. M. P., Vallés, E., Noce, R. D. &
Benedetti, A. V. Novel NiFe/NiFe-LDH composites as competitive
catalysts for clean energy purposes. Appl. Surf. Sci.447,
107116 (2018).
Liu, Z., Sajjad, S. D., Gao, Y., Kaczur, J.
& Masel, R. An Alkaline Water Electrolyzer with Sustainion Membranes:
1 A/cm² at 1.9 V with Base Metal Catalysts. ECS Trans.77,
7173 (2017).
Corrigan, D. A. The Catalysis of the Oxygen
Evolution Reaction by Iron Impurities in Thin Film Nickel Oxide
Electrodes. J. Electrochem. Soc.134, 377384 (1987).
Louie, M. W. & Bell, A. T. An Investigation
of Thin-Film NiFe Oxide Catalysts for the Electrochemical Evolution
of Oxygen. J. Am. Chem. Soc.135, 1232912337 (2013).
Swierk, J. R., Klaus, S., Trotochaud, L.,
Bell, A. T. & Tilley, T. D. Electrochemical Study of the Energetics of
the Oxygen Evolution Reaction at Nickel Iron (Oxy)Hydroxide Catalysts.
J. Phys. Chem. C.119, 1902219029 (2015).
Trotochaud, L., Young, S. L., Ranney, J. K.
& Boettcher, S. W. NickelIron Oxyhydroxide Oxygen-Evolution
Electrocatalysts: The Role of Intentional and Incidental Iron
Incorporation. J. Am. Chem. Soc.136, 67446753 (2014).
Acknowledgements
The authors gratefully acknowledge
support from the Australian Renewable Energy Agency (ARENA), Grant
number DM015 (entitled: Ammonia production from renewables) (G.F.S.
& G.G.W.). This activity received funding from ARENA as part of
ARENAs Research and Development Program-Renewable Hydrogen for
Export. Support from the Australian Research Council Centre of
Excellence Scheme (grant number CE140100012) (G.G.W. and others)
and the Australian National Fabrication Facility (ANFF) Materials
Node is also acknowledged. The authors acknowledge the assistance
of the University of Wollongong Electron Microscopy Centre. This
research used equipment funded by the Australian Research Council
Linkage, Infrastructure, Equipment, and Facilities grant
LE160100063.
Author information
Author notes
These authors contributed equally:
Aaron Hodges, Anh Linh Hoang.
Authors and Affiliations
Intelligent Polymer Research
Institute, University of Wollongong, Wollongong, NSW, 2522,
Australia
Aaron Hodges, Anh Linh Hoang, George
Tsekouras, Klaudia Wagner, Chong-Yong Lee, Gerhard F. Swiegers & Gordon
G. Wallace
Australian Research Council Centre of
Excellence for Electromaterials Research, University of
Wollongong, Wollongong, NSW, 2522, Australia
Klaudia Wagner, Chong-Yong
Lee, Gerhard F. Swiegers & Gordon G. Wallace
Contributions
A.H. and A.L.H. contributed equally
to this work. A.H. developed the model for capillary-induced
electrolyte flow in a porous inter-electrode separator, measured
the ionic resistance of the separators and studied the Faradaic
efficiency and gas crossover of capillary-fed electrolysis cells
employing them. A.H. further developed the model for
diffusion-based gas crossover referred to. A.L.H. developed and
studied all aspects of the electrodes, as reported herein,
including the catalysts, their bubble-free operation, and detailed
optimisation. G.T., K.W., C.Y.L., and G.W. co-supervised or aided
the above work. G.S. was the primary supervisor. The project was
conceived of by G.S. and K.W. G.S. and G.T. wrote and edited the
paper, and all authors commented on it.
G.T., K.W., C.Y.L., and G.S. are
employees of the University of Wollongong but are presently, or
will be in future, performing paid work for Hysata Pty Ltd, a
company that has licensed the capillary-fed electrolysis cell
technology. A.H., A.L.H., K.W., C.Y.L., and G.F.S. are
co-inventors on patent applications covering the technology that
have been filed. The remaining authors declare no competing
interests.
Peer review
Peer review information
Nature Communications thanks
J. Willem Haverkort and Alexandr Oshchepkov for their contribution
to the peer review of this work. Peer reviewer reports are
available.
Additional information
Publishers note Springer Nature
remains neutral with regard to jurisdictional claims in published
maps and institutional affiliations.